文章标题: Long-read metagenomics reveals phage dynamics in the human gut microbiome
中文标题: 宏基因组长读测序揭示人类肠道噬菌体“生存法则”
关键词: 肠道微生物组、噬菌体、长读测序、噬菌体-宿主相互作用、水平基因转移、IScream噬菌体
摘要总结:
这篇文章通过对六名健康个体粪便样本进行为期两年的深度长读宏基因组测序,深入探索了人类肠道微生物组中噬菌体整合动力学。研究团队发现,尽管大多数原噬菌体(prophages)在其细菌宿主中保持稳定整合,但约5%的噬菌体在两年内表现出动态的得失。研究还发现,在同一样本中,同时存在整合了噬菌体的细菌宿主和未整合噬菌体的宿主,表明噬菌体在群体水平上存在异质性。当检测到噬菌体诱导时,其发生率普遍较低(仅为宿主区域覆盖度的1-3倍),这与理论预期一致。研究还识别出多个案例,显示相同的噬菌体整合到不同分类家族的细菌中,挑战了噬菌体宿主特异性的传统观念。此外,文章描述了一类新型的“IScream噬菌体”,它们利用细菌IS30转座酶进行动员,代表了一种此前未被识别的自私细菌元件噬菌体驯化形式。这些发现不仅揭示了人类肠道微生物组中噬菌体-细菌动力学的基本方面,也拓展了我们对驱动水平基因转移和微生物基因组可塑性的演化机制的理解。这对于理解噬菌体在塑造微生物群落和人类健康中的作用具有重要意义。
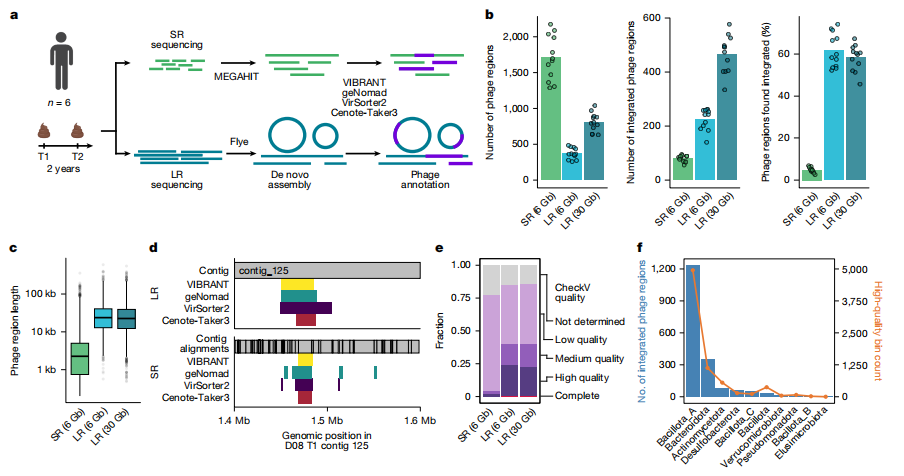
文章的亮点和局限:
文章标题: Assessing phylogenetic confidence at pandemic scales
中文标题: 疫情溯源新利器:SPRTA提升大流行规模系统发育树可信度
关键词: 系统发育学、SARS-CoV-2、基因组流行病学、分支支持度、突变历史、SPRTA
摘要总结:
这篇文章提出了一种名为“子树修剪和再嫁接”的树评估(SPRTA)方法,旨在高效且可解释地评估系统发育树的置信度,特别适用于大流行规模的基因组流行病学数据。传统系统发育支持度方法(如Felsenstein's bootstrap)计算成本高昂且难以解释,尤其是在处理数百万基因组的大流行数据集时。SPRTA通过将系统发育支持度的焦点从评估谱系(clades)的置信度转移到评估演化历史和系统发育位置,从而克服了这些局限性。研究团队将SPRTA应用于超过200万个SARS-CoV-2基因组的全球系统发育树,成功识别了许多变异株可能的替代演化起源,并评估了Pango谱系分类系统的可靠性,同时揭示了系统发育不确定性对推断突变率的影响。结果显示,SPRTA在计算效率上比现有方法快至少两个数量级,并且能够可靠地识别正确推断的突变事件和不确定性较高的突变事件。这些发现为大流行规模的系统发育分析提供了一种新范式,能够更精确地评估病毒传播和突变历史,从而提升我们应对未来大流行的准备和响应能力。这对于基因组流行病学领域,以及理解和应对未来的大流行具有重要意义。
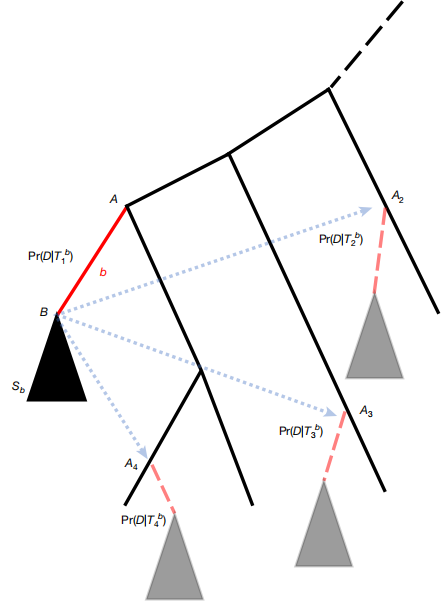
文章的亮点和局限:
文章标题: Estimation and mapping of the missing heritability of human phenotypes
中文标题: 解锁遗传学“黑箱”:全基因组测序精确定位人类表型缺失遗传力
关键词: 缺失遗传力、全基因组测序、稀有变异、常见变异、GWAS、复杂性状
摘要总结:
这篇文章通过对UK Biobank中347,630名欧洲血统个体的全基因组序列(WGS)数据进行分析,深入探索了人类表型缺失遗传力的估计和定位。研究团队量化了4000万个单核苷酸和短插入/缺失变异(次要等位基因频率MAF > 0.01%)对34种复杂性状和疾病遗传力的相对贡献。结果显示,WGS数据平均捕获了约88%的基于家系谱的狭义遗传力,其中20%来自稀有变异(MAF < 1%),68%来自常见变异(MAF ≥ 1%)。研究发现,编码区和非编码区遗传变异分别贡献了稀有变异WGS遗传力的21%和79%。文章识别出15种性状的WGS遗传力与家系谱遗传力估计值无显著差异,表明它们的遗传力已通过WGS数据得到充分解释。此外,通过对所有34种表型进行全基因组关联分析(GWAS),共识别出11,243个常见变异关联和886个稀有变异关联。对于脂质相关性状,超过25%的稀有变异遗传力可被映射到特定基因座,且所需测序样本量少于50万个。这些发现提供了稀有变异遗传力的高精度估计,解释了许多表型的遗传力,并为未来通过GWAS设计更优化的实验以识别复杂性状的因果遗传变异提供了重要指导。这对于理解人类复杂性状的遗传结构,以及改进疾病风险预测和治疗策略具有重要意义。
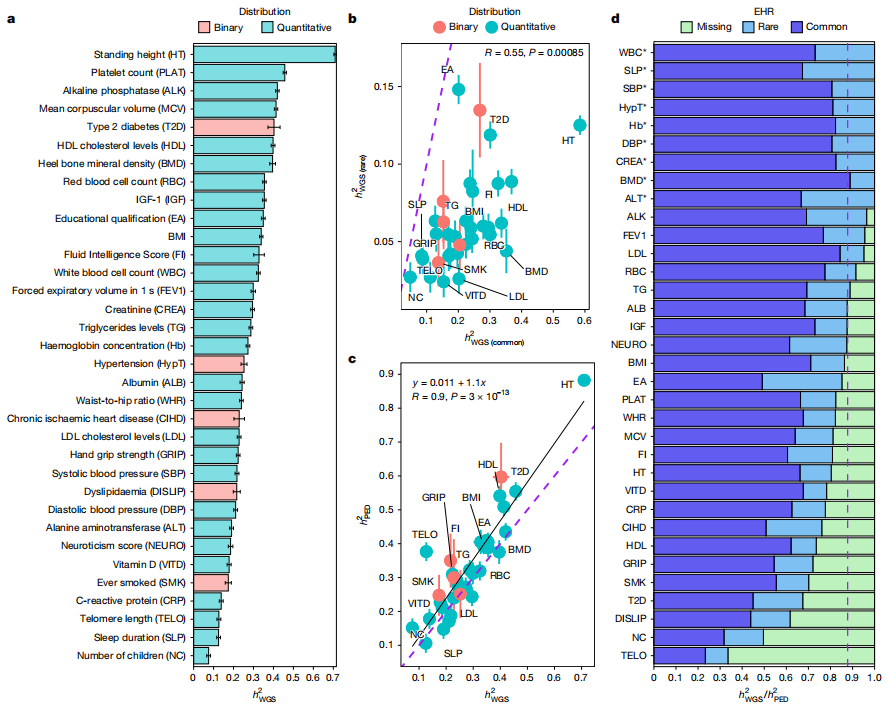
文章的亮点和局限:
文章标题: Ecology and spread of the North American H5N1 epizootic
中文标题: H5N1禽流感北美大流行:候鸟是病毒传播的“超级载体”
关键词: H5N1禽流感、流行病学、野鸟、家禽、传播动力学、系统发育地理学
摘要总结:
这篇文章通过对来自野鸟、家禽和哺乳动物的1,818个血凝素序列进行贝叶斯系统发育地理学分析,深入探索了2021年末至2023年9月北美H5N1高致病性禽流感(HPAI)大流行的生态学和传播模式。研究团队发现,北美大流行是由大约九次独立引入大西洋和太平洋候鸟迁徙路线引发的,随后通过野鸟和候鸟迅速传播。分析表明,雁形目鸟类(如鸭、鹅)是病毒传播的主要驱动因素,而非典型宿主(如猛禽、鸣禽、哺乳动物)主要充当病毒的“终结宿主”。与2015年疫情不同,本次疫情中家禽的暴发是由野鸟重复、独立的引入引发,而非主要通过农场间传播。研究还发现,散养家禽(backyard birds)比商业家禽平均早约9天被感染,提示其可能作为早期预警信号。这些发现明确指出野鸟是H5N1疫情传播的关键驱动因素,强调了加强野鸟监测以及在野鸟-农业界面采取干预措施对于未来疫情追踪和暴发预防至关重要。这对于制定更有效的H5N1防控策略,以及应对未来人畜共患病威胁具有重要意义。
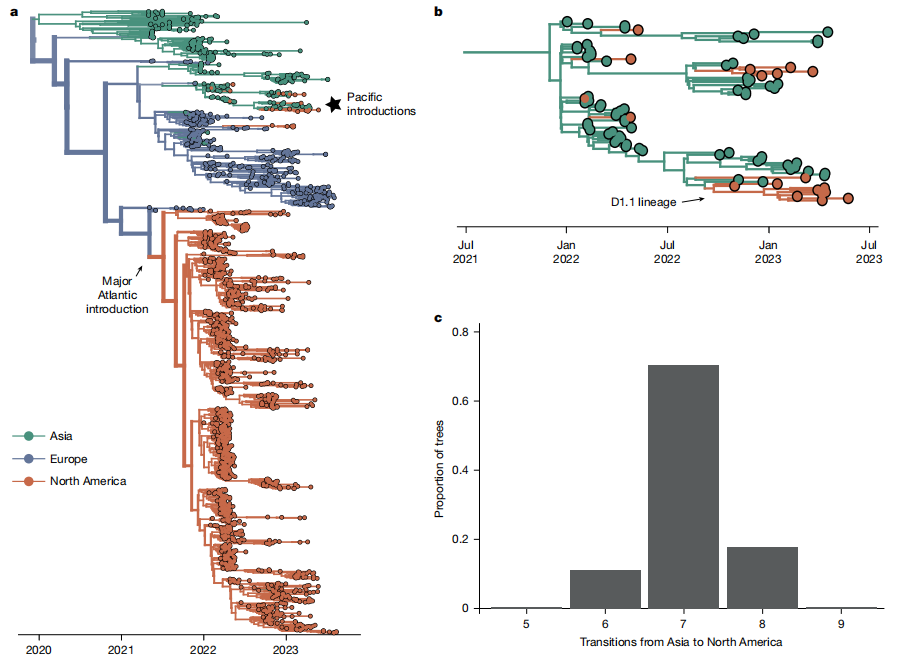
文章的亮点和局限:
致谢橙子牛奶糖(陈文燕),请用参考模版:We thank the blogger (orange_milk_sugar, Wenyan Chen) for XXX
感谢小可爱们多年来的陪伴, 我与你们一起成长~

此内容由惯性聚合(RSS阅读器)自动聚合整理,仅供阅读参考。 原文来自 — 版权归原作者所有。