
Materials design underpins a wide range of technological advances, from nanoelectronics to semiconductor design and energy storage. Yet development cycles for novel materials remain slow and costly. Universal machine learning interatomic potentials aim to accelerate the materials design process by providing accurate stability and property predictions for a wide range of materials. These models are orders of magnitude faster than traditional first-principles simulations, turning previously impractical problems into routine computations that can be completed in a few hours. Since we launched our MatterSim-v1 model, it has gained popularity in the materials science community for its ability to accurately simulate materials under realistic conditions, including finite temperature and pressure.
Today, we have several exciting MatterSim updates to share. These include experimental validation of MatterSim predictions for thermal conductors, performance improvements for faster simulation, and the introduction of a new multi-task foundation model for materials characterization.

Materials with high thermal conductivity play a critical role in heat management, preventing overheating and improving energy efficiency. For example, established high thermal conductors like diamond, copper and silicon are widely used across a broad range of cooling applications. Designing next-generation thermal conductors may enable advances in computing, power electronics, and aerospace technologies. However, doing so requires accurate predictions of thermal conductivity values for candidate materials.
In solids, heat is carried in two main ways: by vibrating atoms (phonons) and by moving electrons. The phonon contribution can be estimated using machine-learning interatomic potentials to enable screening of thousands of candidates, narrowing the search space to the most promising materials before expensive experimental validation.
“MatterSim has generated by far the largest database of computational thermal conductivities. This opens the door to exploring a far broader materials space than before […].”
– Prof. Bing Lv, University of Texas Dallas
In collaboration with the University of Texas Dallas (UT Dallas), University of Illinois Urbana-Champaign, and University of California Davis (UC Davis), we have used MatterSim-v1 to screen over 240,000 candidate materials for high thermal conductors. As shown in Fig. 1 (left), MatterSim’s predictions have good agreement with first-principles simulations. Prof. Davide Donadio from UC Davis: “I was amazed by how the MatterSim model combined accuracy and computational efficiency to predict such a sensitive property as thermal conductivity. That was the key that unlocked screening at this scale, hundreds of thousands of crystals, that would have been completely out of reach with conventional methods.” Prof. Bing Lv from UT Dallas adds: “MatterSim has generated by far the largest database of computational thermal conductivities. This opens the door to exploring a far broader materials space than before, enabling the community to uncover a broader set of viable materials even after imposing practical requirements.”
“For the first time, we can test conventional understanding of what controls thermal conductivity at scale […]”
– Prof. David Cahill, University of Illinois Urbana-Champaign
Based on these predictions, we have identified tetragonal tantalum phosphorus (TaP) as a potential high thermal conductor. We have experimentally synthesized tetragonal tantalum phosphorus (TaP) at UT Dallas and measured its thermal conductivity at University of Illinois Urbana-Champaign (152 W/m/K for our best samples), close to the thermal conductivity of silicon. While we are not the first to synthesize tetragonal TaP, the material has not been considered as a thermal conductor before. These results demonstrate how MatterSim can enable the identification of functional materials: “For the first time, we can test conventional understanding of what controls thermal conductivity at scale, while enabling the discovery of new functional materials that balance it with other important constraints such as mass density, elemental abundance, and environmental stability”, says Prof. David Cahill from University of Illinois Urbana-Champaign.
video series

A video series with Sinead Bovell built around the questions everyone’s asking about AI. With expert voices from across Microsoft, we break down the tension and promise of this rapidly changing technology, exploring what’s evolving and what’s possible.
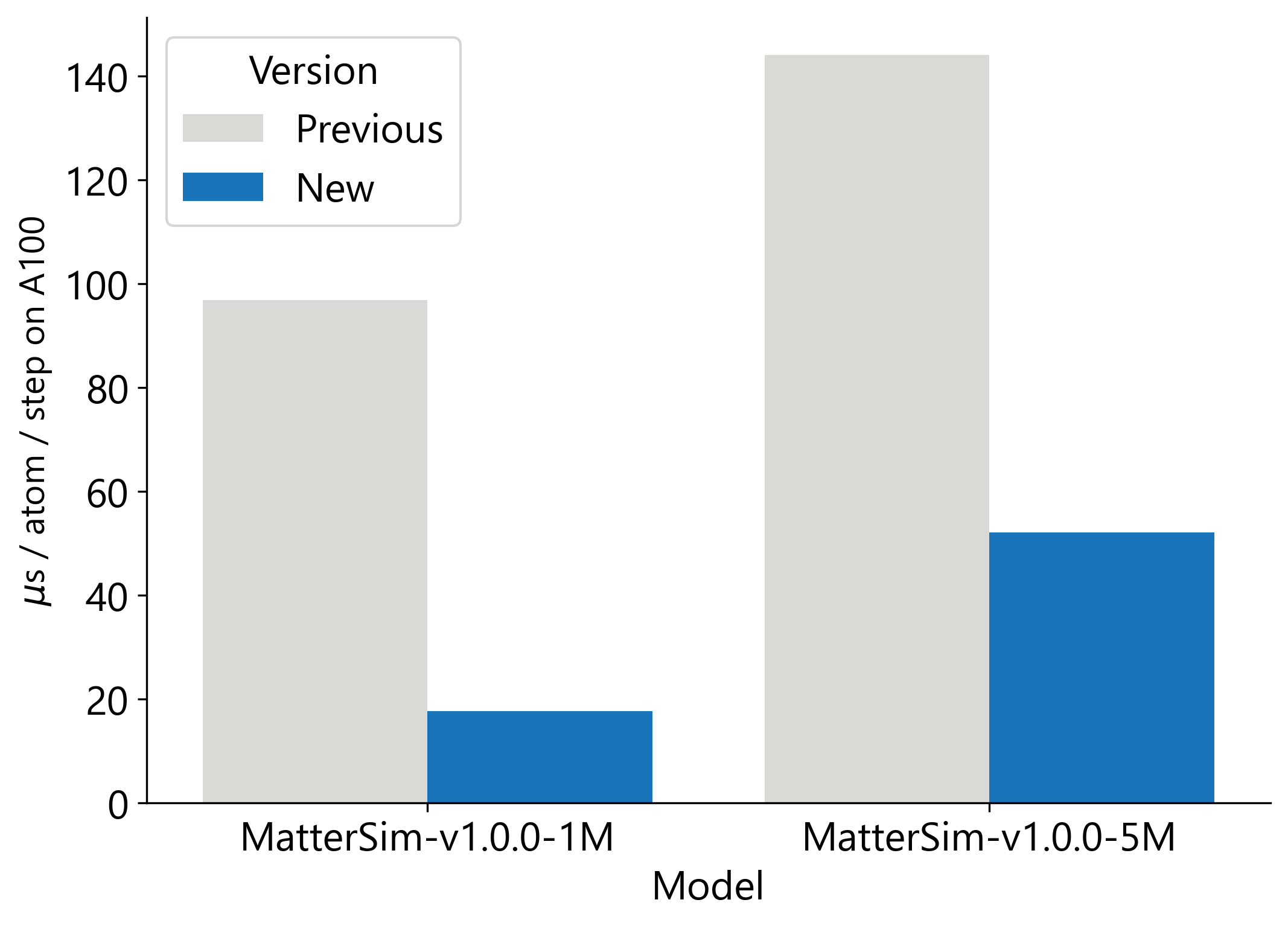
We are making MatterSim-v1 significantly faster by releasing several open-source performance and usability improvements. First, we speed up model inference through a combination of faster graph construction, ahead-of-time compilation and reduced conversion between atomic representations, resulting in a 3x speed-up of MatterSim-v1.0.0-5M and a 5x speed-up of MatterSim-v1.0.0-1M (see Fig. 2). To make MatterSim-v1 easier to use, we have integrated it into the widely used LAMMPS simulation software, allowing users to easily scale model inference across multiple GPUs in their existing workflows.
Building on the success of MatterSim-v1, today we extend the MatterSim model family by announcing MatterSim-MT: a multi-task (MT) foundation model for in silico materials simulation and property characterization. The model natively predicts energies, forces, stress and several important materials properties.
MatterSim-MT is pretrained on over 35 million first-principles-labelled structures covering 89 elements, temperatures up to 5000 K and pressures up to 1000 GPa. It is further fine-tuned on various properties including Bader charges, magnetic moments, Born effective charges, and dielectric matrices. Out of the box, MatterSim-MT serves as a foundation model for predicting material structure, dynamics and thermodynamics. Its multi-task architecture also enables a wide range of complex simulations that cannot be captured by potential energy surfaces alone. The ability to accurately simulate these phenomena is crucial for applications such as catalysis and energy storage.
Here, we illustrate these multi-task capabilities through three case studies: vibrational spectroscopy, ferroelectric switching, and electrochemical redox. Each example requires a distinct combination of property predictions. In the full manuscript, we also show that MatterSim-MT scales well with more data and parameters, can be efficiently fine-tuned to higher levels of theory, and can be systematically extended to new systems via active learning.
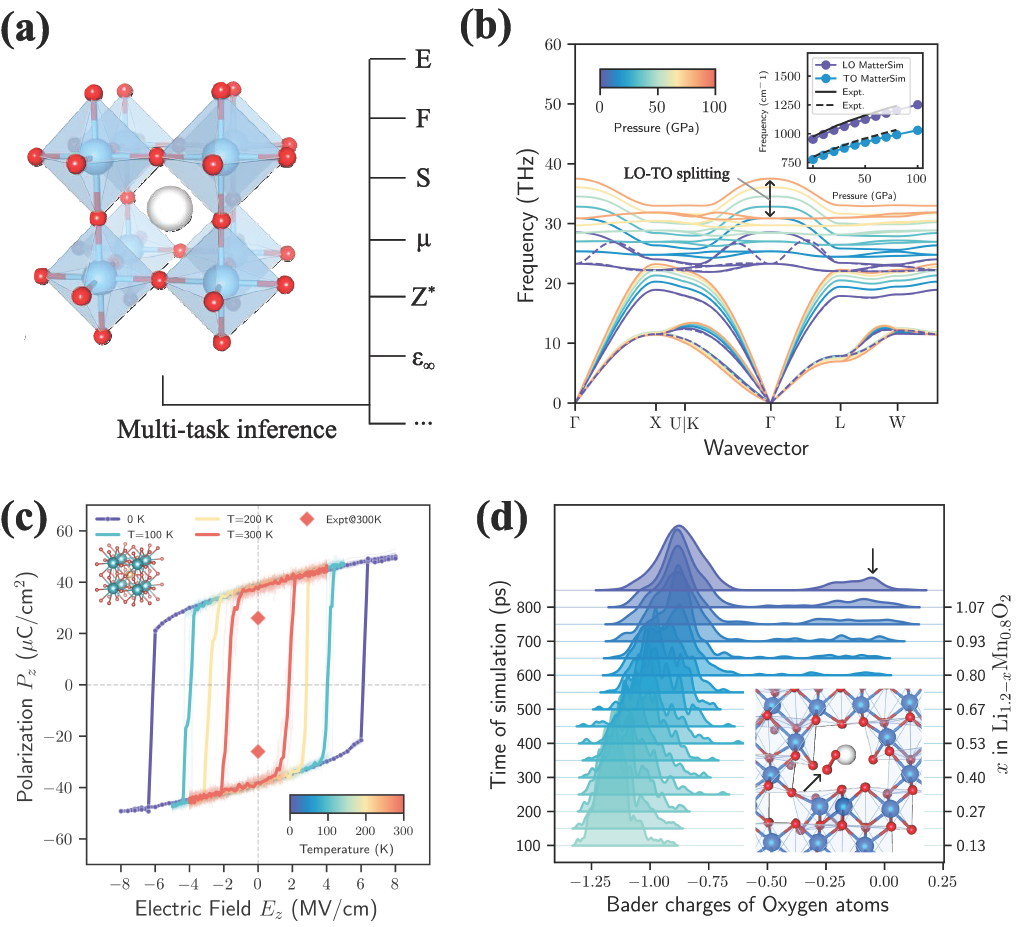
First, we focus on vibrational spectroscopy, a technique that identifies substances by measuring how their atomic bonds naturally vibrate. We demonstrate how predictions of Born effective charges and dielectric properties enable the computation of phonon spectra in polar crystals. In these materials, oppositely charged ions vibrate against each other. Depending on the direction of vibration, this can lead to a buildup of charge that creates a macroscopic electric field, splitting the optical phonon modes into higher-frequency longitudinal (LO) and lower-frequency transverse (TO) branches. As a case study, we simulated this behavior in 3c-silicon carbide (3c-SiC), a material used in high-power electronics, under extreme pressures. As shown in Fig. 3(b), MatterSim-MT predicts a Born effective charge in close agreement with both theoretical and experimental values. The resulting LO-TO splitting of 5.26 THz deviates by only 0.06 THz from ab initio calculations and 0.03 THz from experimental measurements.
The predicted Born effective charges also allow us to simulate how systems respond to an external electric field. In ferroelectric materials, ions adopt an asymmetric arrangement that gives the crystal a net electric polarization that can be flipped by an applied field. In Fig. 3(c), we demonstrate this by simulating barium titanate (BaTiO3) under an applied electric field, reproducing the switching of its polarization. The resulting hysteresis curve correctly shows that finite-temperature effects at 300 K make it easier to flip the polarization, even though the predicted spontaneous polarization (38 μC/cm2) is slightly higher than the experimental value (26 μC/cm2). This discrepancy is likely due to the well-known underbinding of the underlying first-principles calculations.
Finally, we predict atomic charges to study the electronic degrees of freedom in chemical bonding and redox processes. We examine the behavior of the cathode material Li1.2 – xMn0.8O2 during a simulated battery charging process. These lithium-rich transition-metal oxides are promising next-generation batteries due to their high energy density but suffer from irreversible capacity loss associated with the anionic oxygen redox mechanism. We reproduced this phenomenon by running molecular dynamics simulations at 1000 K and progressively extracting Lithium to mimic battery charging. We observe a clear shift over time: at first, the manganese (Mn) atoms supply the electrons needed for charging, but as more lithium is removed, oxygen atoms are forced to give up electrons instead (cationic to anionic redox), as shown by the shift to less negative Bader charges over time (Fig. 3(d)). This destabilises the structure with oxygen atoms pairing up to form O2 dimers (Fig. 3(d), inset). Notably, this comprehensive picture of the cationic-to-anionic redox transition and lattice degradation naturally emerges from the multi-task predictions, without any task-specific training on battery materials.
With experimental validation, substantial performance improvements, and new multi-task capabilities, MatterSim is advancing toward more practical, decision-relevant use in materials design. Together, these developments are helping materials scientists move more quickly from large-scale computational screening to targeted experimental follow-up and decision-relevant scientific workflows. We are excited to see how the materials science community applies these advances in their own domains.
We look forward to continued collaboration as MatterSim is tested, extended, and integrated into real-world materials discovery pipelines.
This work is the product of a highly collaborative and interdisciplinary effort led by Microsoft Research AI for Science in partnership with Microsoft Research Accelerator and collaborators at the University of Texas Dallas (supported by MSR Accelerator), University of Illinois Urbana-Champaign and University of California Davis. Contributors to this work include Han Yang, Xixian Liu, Chenxi Hu, Yichi Zhou, Yu Shi, Chang Liu, Junfu Tan, Jielan Li, Guanzhi Li, Qian Wang, Yu Zhu, Zekun Chen, Shuizhou Chen, Fabian Thiemann, Claudio Zeni, Matthew Horton, Robert Pinsler, Andrew Fowler, Daniel Zügner, Tian Xie, Lixin Sun, Yicheng Chen, Lingyu Kong, Yeqi Bai, Deniz Gunceler, Frank Noé, Hongxia Hao, Ziheng Lu, Zixin Zhai, Mengfan Wu, Haoke Qiu, Mingfa Tang, Tie-Yan Liu, Haiguang Liu, Tao Qin, David G. Cahill, Bing Lv, Davide Donadio, Shoko Ueda, and Kenji Takeda.
此内容由惯性聚合(RSS阅读器)自动聚合整理,仅供阅读参考。 原文来自 — 版权归原作者所有。